
性别:女性
一般病史:
患者女性,34岁,发现头部肿物20余天。患者20余天前发现枕部按压痛,半月来出现枕部头皮肿胀,直径约2cm,与周围组织分界不清,按压疼痛,伴有头痛。药物治疗后头痛较前减轻,停药后复发。CT检查示右侧顶骨占位性病变。MRI平扫:右侧顶骨孤立性占位。MRI增强:右侧顶骨占位并周围头皮软组织肿胀,结合病史及MR+CT检查,考虑骨髓炎。
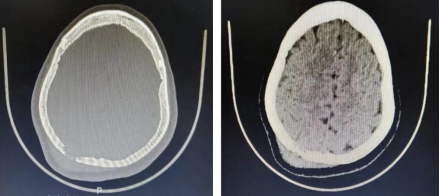
图1 CT:右侧顶骨可见一丘状稍高密度影,最大截面约40mm×25mm,病变呈膨胀性生长,邻近顶叶稍受压,颅板骨质局部破坏。
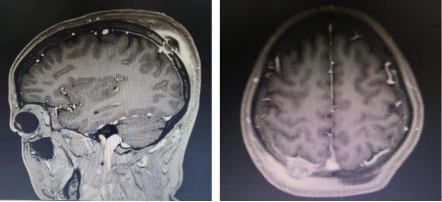
图2 MRI:右侧顶骨结节状异常信号,DWI呈不均高信号,增强呈不均强化,邻近脑膜呈线样强化,邻近头皮软组织肿胀并明显均匀强化。
病理学检查
大体:灰红组织一块,大小4.2cm×4cm×1.7cm,表面有破溃,破溃处大小2.5cm×1.5cm,切面灰红灰白质中,另见灰白骨样组织多块,大小2cm×2cm×1cm。
镜下特点
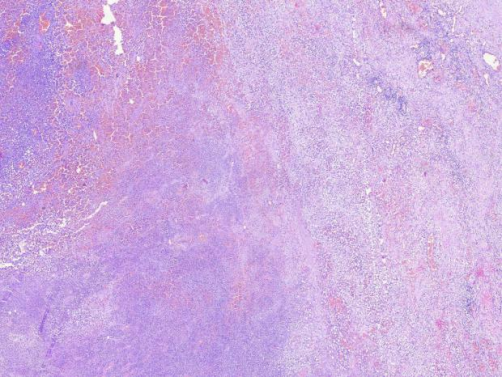
图3 低倍镜下,病变呈弥漫性分布,局部区域染色较深,可见出血
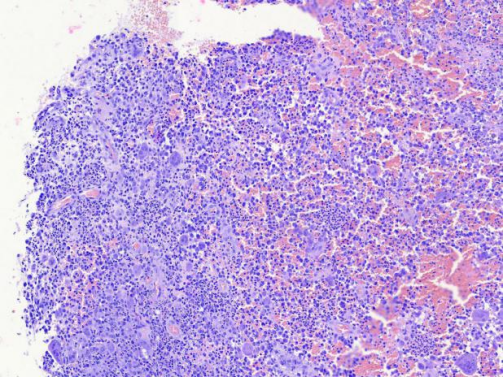
图4 病变细胞弥漫性分布,细胞体积较大,可见多核细胞,病变内弥漫性炎细胞浸润,局部有出血
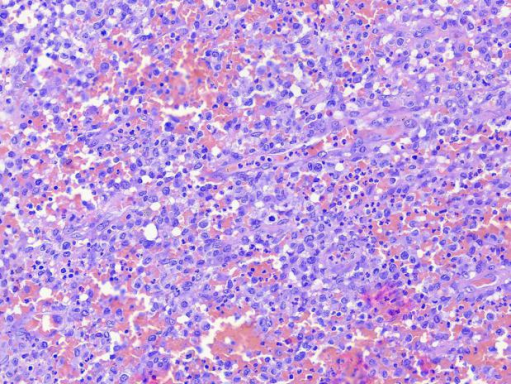
图5 中倍镜下,病变细胞呈卵圆形,体积中等至较大,细胞核肾形有偏位,可见核分裂象。病变内可见较多中性粒细胞
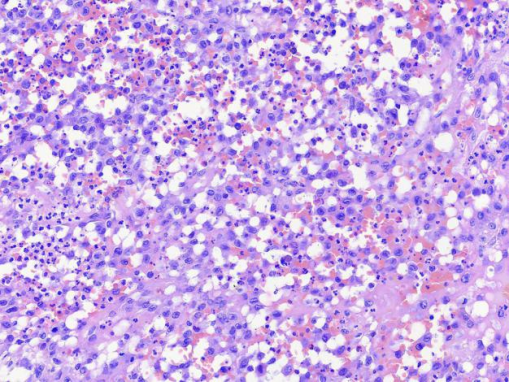
图6 高倍镜下,病变细胞弥漫性分布,细胞卵圆形或不规则形,细胞核偏位,可见核沟。背景细胞中含有小淋巴细胞、中性粒细胞、嗜酸性粒细胞。
免疫组化结果:病变细胞弥漫性表达S-100、CD1α、Langerin和CD68
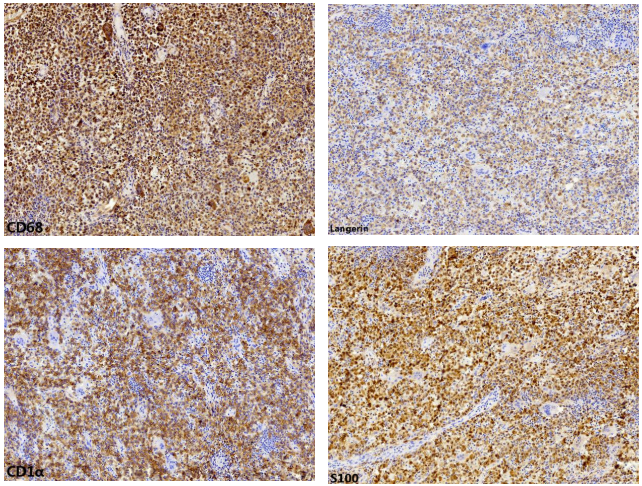
图7 朗格汉斯组织细胞增生症的免疫组化特征
病理诊断:(顶骨)朗格汉斯组织细胞增生症,侵犯周围骨质。
讨论:朗格汉斯组织细胞增生症(LCH)是一种免疫组化表达CD1a、Langerin和S100蛋白;超微检查中显示Birbeck颗粒的朗格汉斯型细胞的克隆性肿瘤性增生。
临床特点:
1.多见于儿童(1-3岁),成人罕见
2.男性多见,男女比例为3.7:1
3.多见于北欧后裔,黑色人种罕见
4.可分为单灶性、单系统多灶性和多系统多灶性病变。
1)单灶性:主要位于骨和邻近软组织(颅骨、股骨、椎体及骨盆),少见情况下可为淋巴结、皮肤和肺。通常为年长儿童或成人,最常表现为溶骨性骨病变,侵蚀皮质。
2)单系统多灶性:最常累及骨骼系统,主要为颅骨和下颌骨。病灶可从2 个到4个或更多,其次为淋巴结、皮肤、女性生殖系统、消化系统和肺。多见于较年轻儿童,临床症状主要取决于受累的脏器。
3)多系统多灶性:主要为婴儿,临床上常有广泛的红斑样鳞屑或湿疹样皮疹、发热、肝、脾和淋巴结肿大;多发性溶骨性损害、发热、贫血和血小板减少等。
影像学表现:
早期为溶骨性破坏,边缘模糊,可出现骨膜反应。晚期边缘清晰锐利,出现硬化。
组织学特点:
1.肿瘤背景细胞混杂,可见中性粒细胞、淋巴细胞、嗜酸性粒细胞,也可见多核巨细胞。
2.肿瘤细胞,典型的LCH细胞胞浆中等量,粉染,可见核沟或卷曲状外形;染色质常呈空泡状,核仁不明显,核异型性较小,分裂像可见或少见。
免疫组化:
1)持续表达CD1α,Langerin和 S-100。
2)可表达vimentin、CD68和HLA-DR。
3)CD45和溶菌酶呈低表达。
4)不表达B细胞和T细胞谱系标记物(CD4除外)、CD30和滤泡辅助性细胞标志物
5)Ki67增殖指数高度不一
分子遗传学:
1)约30%病例有克隆性IGH、IGK或TR重排
2)约50%病例存在BRAF V600基因突变。
鉴别诊断:
1.淋巴瘤(霍奇金淋巴瘤、间变大细胞淋巴瘤) 霍奇金淋巴瘤和间变性大细胞性淋巴瘤中的R-S细胞和hallmark细胞形态学比较类似于LC,但R-S细胞表达表达CD30、CD15;间变性大细胞性淋巴瘤细胞达T细胞性标记、ALK和CD30,不表达S100、CD1a和Langerin。
2.Kimura病:多见于青年男性,好发于淋巴结(颈部尤以腮腺和枕顶部淋巴结)及其邻近软组织。组织学表现为淋巴结结构基本保存,淋巴滤泡增生,生发中心扩大,多量嗜酸性粒细胞弥漫或灶性浸润,少量组织细胞增生。免疫组化组织细胞表达CD68,不表达CD1a和S-100。
3.骨髓炎:当LCH发生于骨骼而且伴发较多的中性粒细胞、淋巴细胞时可类似于骨髓炎。但骨髓炎中还可以出现浆细胞,这罕见于LCH中。另外,骨髓炎中增生的组织细胞不表达S100、CD1a和Langerin。
4.Rosai-Dorfman病:表现为双侧颈部淋巴结无痛性肿块,伴发热、血沉增快等。镜下突出的形态为窦内充满组织细胞及少量小淋巴细胞,组织细胞有明显的吞噬淋巴细胞等现象。免疫组化组织细胞虽然表达S-100,但不表达CD1a和Langerin。
5.树突细胞肿瘤:病变细胞呈梭形或圆形,排列成束状或漩涡状,瘤细胞除了表达S-100外,还表达CD21、CD35等标记可有助于鉴别。
治疗和预后
LCH治疗根据病变发生部位和受累器官损伤程度来决定。如果病变能够手术,则应进行病灶完整切除;若只能部分切除时,应进行局部放疗。多病灶、多器官受累者,应该进行全身系统治疗。
多数单一病灶LCH患者预后较好,而2个或2个以上部位受累的患者生存率明显下降,出现肝、脾、肺及骨髓等器官受累时提示预后较差。
参考文献:
1.Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of
haematopoietic and lymphoid tissues. Revised 4th Edition, IARC, 2017.
2.Jaffe ES, Harris NL, Vardiman JW, et al. Hematopathology. Saunders Elsevier. 2nd.
3.El Demellawy D, Young JL, de Nanassy J, Langerhans cell histiocytosis: a
comprehensive review. Pathology. 2015,47(4):294-301.
4.龚西騟. 郎格汉斯细胞组织细胞增生症的病理与临床。临床与实验病理学杂志,2000,16(2):149-153.
5.葛荣,殷宪刚,刘创峰,等. 成人朗格汉斯细胞组织细胞增生症的临床病理学分析。中华医学杂志,2012,92(42):2995-2997.


 苏公网安备 32011402010112号
苏公网安备 32011402010112号