

急性淋巴细胞白血病(acute lymphoblastic leukemia,ALL),是一种常见的恶性血液病,生物学特征多样而临床异质性很大,以骨髓和淋巴组织中不成熟淋巴细胞的异常增殖和聚集为特点。ALL占所有白血病的15%,约占急性白血病的30%~40%。发病率在美国白人中为1.5/10万,黑人为0.8/10万;男女之比为1.4:1。我国1986年白血病流行病学调查研究显示我国的ALL发病率为0.69/10万。美国统计资料显示75%的患者<15岁,发病高峰在3~7岁,10岁以后发病率随年龄增长逐渐下降,但50岁以后发病率又略有上升。成人ALL的中位年龄30~40岁。通常男性比女性稍多见。ALL包括B-ALL及T-ALL,其中B-ALL中20%~30%患者染色体伴(9;22)(q34;q11.2)/BCR-ABL1重现性遗传学异常,称为Ph+ALL。
二、临床表现
急性白血病的临床表现包括骨髓组织受白血病细胞浸润所引起的骨髓正常造血衰竭表现(如贫血、感染、出血等)以及白血病细胞的髓外浸润引起的异常(如淋巴结、肝脾肿大等)两大方面。ALL的临床表现各异,症状可以表现比较隐匿,也可以呈急性,这取决于骨髓被恶性克隆替代的程度和髓外浸润的范围;患者就医前的症状期平均约6周(可短于1周至长达1年)。与急性髓系白血病比较,起病情况及发热、出血、贫血等症状基本相似,但ALL的髓外浸润及中枢神经系统白血病更常见。
(一)正常骨髓造血功能受抑制的表现
1.贫血
贫血是白血病最常见的症状之一,常较早出现,且随着病情进展而加重。表现为苍白、无力、头晕、心悸、厌食、浮肿等。患者贫血的程度与出血量不成比例。
2.出血
出血也是常见表现,约半数病例可有不同程度出血。出血部位分布广泛,以皮肤、黏膜最常见,表现为皮肤瘀点、瘀斑及鼻出血、齿龈出血等。颅内出血、消化道出血、泌尿系出血虽少见,但往往导致严重后果。血小板质和量的异常是出血的最主要因素。白血病细胞对血管壁浸润破坏也增加出血风险。弥散性血管内凝血(DIC)的发生、凝血因子缺乏也可加重出血倾向。
3.发热、感染
一半以上患者由发热起病,可为低热或高热。疾病本身可以出现肿瘤热,但高热往往提示有继发感染。感染可发生于机体任何部位,以咽炎、口腔炎最多见;上呼吸道及肺部感染、肛周感染和胃肠炎也较常见;若合并脓毒血症是引起死亡的主要原因之一。
(二)白血病细胞增殖浸润的表现
白血病细胞可以浸润任何器官,其中淋巴结、肝、脾、骨关节、中枢神经系统和皮肤是最容易受累及的部位。
1.肝、脾、淋巴结肿大:以轻、中度肝脾肿大多见。ALL患者肝脾肿大的发生率较急性髓系白血病发生率高,肿大程度也更明显。淋巴结肿大多见,约50%病例诊断时可发现淋巴结肿大,可累及浅表或深部如纵膈、肠系膜、腹膜后等淋巴结。肝、脾、淋巴结肿大程度一般在T-ALL较B-ALL明显。
2.骨关节疼痛:骨和骨膜的白血病浸润引起骨痛(儿童较成人多见、ALL较急性髓系白血病多见),骨痛常比较剧烈,部位不固定,主要见于四肢骨、脊柱和骨盆,游走性不明显,应用一般止痛剂疗效不佳。逾1/3的患者有胸骨压痛,是白血病常见的体征之一(有助于诊断)。此外,少数患者可因骨髓坏死而导致剧烈骨痛。
3.中枢神经系统白血病(central nervous system leukemia CNSL):CNSL多发生在白血病的缓解期,初诊病例相对少见;ALL的CNSL发生率比在急性髓系白血病(AML)高。浸润部位多发生于蛛网膜、硬脑膜,其次为脑实质、脉络膜或颅神经。CNSL可影响脑脊液(CSF)循环,造成颅内压增高,患者出现头痛、恶心、呕吐、视力模糊、视乳头水肿,甚至抽搐、昏迷等表现。颅神经麻痹主要为神经根被浸润,特别是通过颅神经孔处的第3对和第7对颅神经受累,可引起面瘫;脊髓受白血病细胞浸润,以进行性截瘫为主要特征;血管内皮受浸润以及白血病细胞淤滞,发生继发性出血,临床表现同脑血管意外。
4.睾丸:睾丸白血病是仅次于CNSL的白血病髓外复发的根源,也常出现在缓解期的ALL患者。主要表现为睾丸无痛性肿大,质地坚硬无触痛;多为一侧性,另一侧虽无肿大,但在活检时往往也发现有白血病细胞浸润。明确诊断需要病理活检。
白血病浸润还可累及肺、胸膜、肾、消化道、心、脑、子宫、卵巢、乳房、腮腺和眼部等各种组织和器官,并出现相应脏器的功能障碍,但也可无症状表现。
三、诊断分型
20世纪70年代之前细胞形态学、细胞化学是唯一的诊断工具,此后逐渐发展为:细胞形态学、细胞化学、细胞遗传学(常规细胞遗传学)、免疫表型(多参数流式细胞仪-MFC)、分子细胞遗传学[荧光原位杂交(FISH)],比较基因组杂交技术)、分子遗传学(大多数是以聚合酶链反应—PCR为基础的技术和测序),以及免疫球蛋白和T细胞受体基因重排、多药耐药、基因组学、微小残留病(MRD)等。因此,ALL的诊断分型是一个多步骤的过程,ALL的现代检查、诊断方法应包括精确的免疫学、细胞遗传学和分子生物学。这些方法的结合有助于确定预后相关因素、微小残留病的检测标记,有针对性设计治疗策略。
(一)细胞形态学
ALL分型主要有FAB(French-American-British)和WHO(World Health Organization)两种标准。其中FAB标准主要是以细胞形态学为基础的,要求骨髓中原始淋巴细胞比例超过30%。
法国、美国、英国(FAB)协作组于1976年用Romanowsky染色观察血片及骨髓涂片,根据细胞大小、核浆比例、核仁大小及数量、细胞浆嗜碱程度等,将ALL分为L1、L2、L3三个亚型(表1)。

细胞化学染色弥补了形态学的部分不足,在一定程度上提高了诊断的准确度。ALL患者细胞化学染色的特点主要为:(1)过氧化物酶(POX)与苏丹黑染色(SBB),各阶段淋巴细胞均为阴性,阳性的原始细胞<3%;(2)糖原染色(PAS),约20%~80%的原始淋巴细胞呈阳性反应,为红色颗粒状、块状或呈环状排列,胞质背景清晰;(3)酸性磷酸酶染色,T细胞阳性,B细胞阴性;(4)a-丁酸萘酚酯酶(a-NBE)染色呈阴性反应。
(二)免疫分型
免疫表型分析从早期的间接荧光法发展到目前的多色流式细胞术,可以根据细胞大小、颗粒、抗原表达特征将细胞分为不同的群体。
ALL患者的免疫表型分析不仅可以确定受累的系列(B或T细胞系),还可以进一步分析临床重要的亚型,是ALL分型最为重要的检查之一。分析免疫表型时还应注意白血病细胞抗原表达的强度,具体体现在荧光强度的不同(意义在于从正常细胞中分离出白血病细胞、区分不同的白血病亚型)。MFC可以确定绝大多数患者的白血病相关的免疫表型,主要依据为:①交叉系列标记的不同步表达;②某些抗原表达的缺失;③抗原表达的不同步性;④抗原的过表达。因此,幼稚细胞的免疫表型研究包括:①系列确定;②评估细胞成熟情况;③异常表型分析等几方面内容。
白血病细胞群抗原表达强弱的确定还有一定的治疗意义(为单克隆抗体的临床应用提供依据)。定量流式细胞仪分析有助于分析白血病细胞抗原结合位点,对于诊断和MRD监测意义重大。
因此,免疫分型是确诊ALL的重要手段,也是治疗后疾病监测(如MRD)的极有价值的工具。要达到这一目的需要一系列的抗体,可以根据抗原的系列特异性分步筛选。
第1轮筛选:
B淋巴:CD19、胞质CD22、CD79a、CD10
T淋巴:胞质CD3、CD2、CD7
髓系:抗MPO、CD13、CD33、CDw65、CD117
非系列特异性:TdT、CD34、HLA-DR
第2轮筛选:
B-ALL:胞质IgM、k、l、CD20、CD24
T-ALL:CD1a、膜CD3、CD4、CD5、CD8、抗TCRa/b、抗TCRg/d
AML:抗溶酶体、CD14、CD15、CD41、CD61、CD64、抗糖蛋白A。
1994年在法国召开了欧洲白血病免疫学分型协作组(EGIL)会议,提出ALL的四型21类法。即先按T、B淋巴细胞系和髓系抗原积分系统确定不同抗原积分,再按积分和抗原表达及分化程度把ALL分为四大类型(裸型、纯型、变异型、多表型)、21亚型。1995年发表了简化后的EGIL分型,1998年又进行了修改(表2)。
在此基础上99%的病例可以确诊。成人ALL中B-ALL占75%,T-ALL占25%,约25%~30%的成人ALL表达髓系相关抗原。

(三)细胞遗传学和分子学分析
细胞遗传学和分子学分析对于ALL的诊断和预后因素的确定均十分重要,主要涉及染色体易位、缺失、相应的受累基因,细胞周期调控基因等。方法学包括染色体核型分析的常规细胞遗传学、FISH、比较基因组(CGH)、光谱核型分析,分子学分析的聚合酶链反应(PCR,尤其是实时定量PCR)。这些技术的应用可以发现ALL患者的染色体和分子缺陷,可以从分子-遗传学角度对ALL进行分类,获得有益的预后判断资料,为预后分组、分层治疗提供生物学基础。
约60-80%的B-ALL和35-60%的T-ALL有染色体核型异常。ALL患者的染色体核型异常分为倍体异常和结构异常。
倍体异常指染色体数量的异常。超二倍体核型指染色体数量>46;高超二倍体指染色体数量>50,往往提示较好的预后。超二倍体往往指出现额外的4、6、10、14、18和21号染色体等。染色体数目<46条为亚二倍体核型,亚二倍体预后不良,特别是低亚二倍体组(32~39条;90%以上的患者伴有TP53突变)。
结构异常最常见的是平衡易位,平衡易位常导致交叉基因的融合。这些基因重排常与不同的免疫学亚型有关,在儿童和成人ALL中的发生率不一样(表3)。成人ALL最常见的细胞遗传学异常是Ph染色体的异常,即[t(9;22)/BCR-ABL1];发生率可由儿童的5%至老年患者的40%。Ph染色体常见于前体B-ALL,免疫表型常同时表达异常的髓系抗原。少数患者BCR-ABL1重排呈隐匿性,即染色体分带技术无法发现,间期FISH(IP-FISH)和(或)RT-PCR(反转录PCR)可以发现。
细胞遗传学异常是ALL患者的一个标志,对ALL分类和危险度分层至关重要(成人ALL的细胞遗传学预后分组见表4)。但约30%的儿童ALL和50%的成人ALL缺少与临床相关的细胞遗传学异常,基因芯片技术的开展在一定程度上弥补了这一缺陷。以DNA基因芯片为基础的实验除了了解分类诊断、明确患者的分子学特征外,还可以确定与特殊的分子异常、肿瘤表型、临床结果相关的基因表达类型。
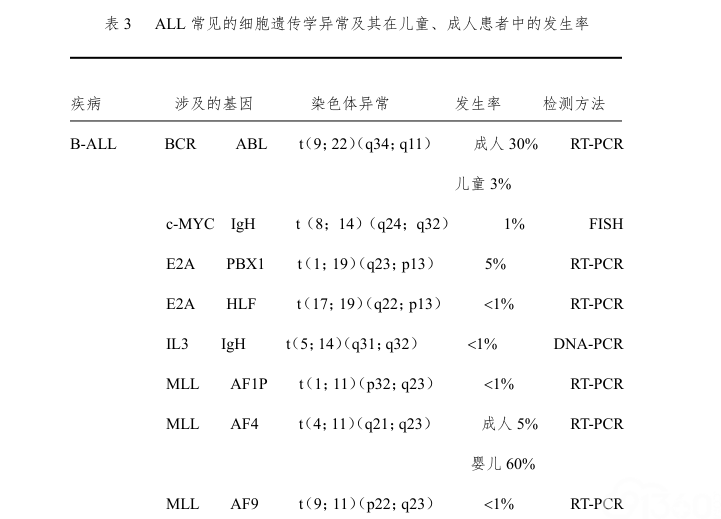
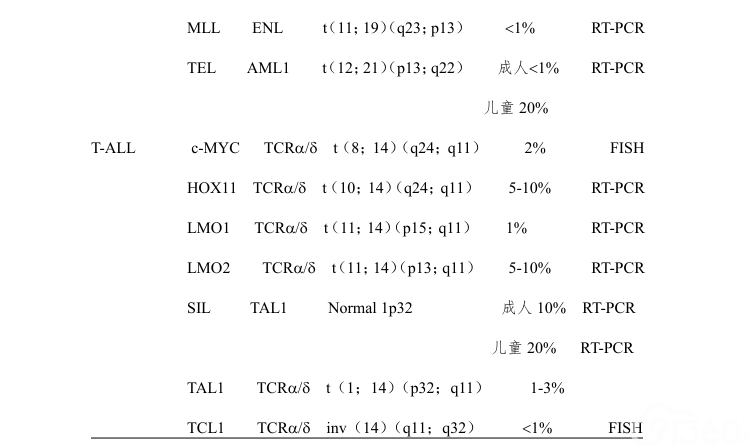

(四)ALL的形态学、免疫学、细胞遗传学、基因分型(MICM分型)
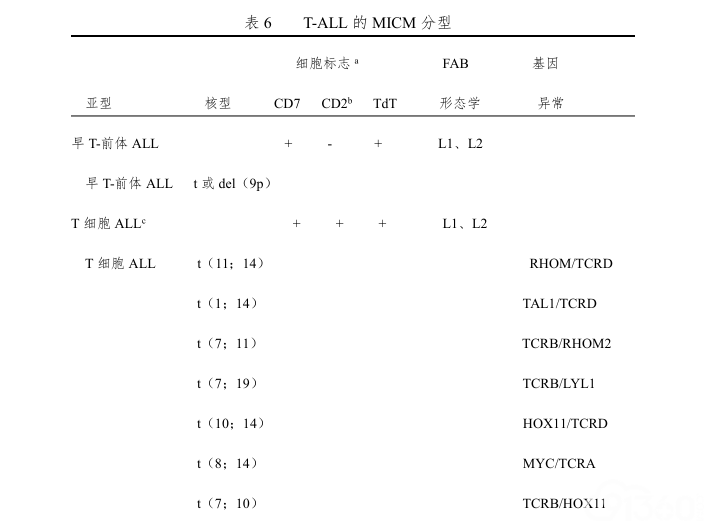
1985年4月由Van den Bergh等在比利时组成了第一个MIC(形态学、免疫学、细胞遗传学)研究协作组,讨论并制定了ALL的MIC分型。高分辨染色体分带技术及分子生物学技术的应用,使ALL分型又前进了一步,出现了MICM分型(形态学、免疫学、细胞遗传学及基因分型,表5、6)。它对于判断预后、指导治疗及微量残留白血病细胞的检测有重要意义。



(五)WHO分型
急性白血病(acute leukemia AL)的高度异质性客观上要求诊断和分型应该综合考虑病因、致病机制、临床表现、细胞形态、免疫表型、遗传学特征、治疗和预后等各种疾病要素。1995年至1997年,世界卫生组织(WHO)召集世界各地著名的临床血液学家和病理学家,在修订的欧洲-美国淋巴组织肿瘤分类(revised European and American classification of lymphoid neoplasms,REAL)的基础上,共同制定了包括AL在内的造血和淋巴组织肿瘤的诊断分型标准,并于2001年正式发表。WHO诊断分型标准突出了细胞分子遗传学异常在疾病诊断和分型中的作用,结合病史、形态、细胞化学和免疫表型等来界定病种。2008年又做了修订。但自2008年WHO更新造血与淋巴组织肿瘤分类后,许多与AL相关的独特生物标志物相继被发现,这些生物标志物绝大部分来源于基因表达分析和二代测序,显著地改善了WHO分类中亚型的诊断标准以及与预后的相关性。于是在2014年春,由100位国际病理学家、血液病学家、肿瘤科医师和遗传学家组成的临床顾问委员会提出了新的修改意见。修订版仍遵循旧分类的原则,按形态学、免疫表型、细胞遗传学和分子基因来定义具有临床意义的独立病种。因此,2016年WHO造血与淋巴组织肿瘤分类仅是对原有类型做了必要的修正和补充,增加了近年来被认识和明确的新类型。下面将详细介绍2016版AL的WHO诊断分型及各分类亚型的形态学、免疫表型和遗传学特征(表7)。

注:1)a为新增加分型。 2)ALL=B淋巴母细胞白血病。
1.ALL,非特指型
B-ALL患者有贫血、中性粒细胞和血小板减少,常见肝、脾和淋巴结肿大。儿童B-ALL的CR率>95%,治愈率约为80%,但成人CR率较低,仅为60%~85%,治愈率<80%;强化疗可以提高年轻ALL患者的治愈率。婴幼儿或年龄>10岁、WBC数高、浸润者诱导治疗预后不佳。
(1)形态学:细胞体积小者,胞质稀少,核染色质凝聚,核仁不明显;体积大的胞质量中等,淡蓝或灰蓝色;核染色质弥散,可有多个明显的核仁。有的胞质有伪足,称“手镜细胞”。细胞化学染色在ALL诊断中的价值不如在AML大。原始淋巴细胞MPO阴性。如果胞质有颗粒,SBB染色可以呈淡灰色,强度不及AML。PAS阳性,通常为粗大颗粒或呈块状。NSE染色在胞质中呈多点状分布,或位于高尔基复合体区,氟化钠抑制程度不一。
(2)免疫表型:表达B细胞标记CD19、cCD79a和cCD22,但其中单独一个阳性不能认定为B细胞。如果荧光强度高,则有利于B细胞来源的判断。多数患者原始淋巴细胞表达CD10、sCD22、cCD24,PAX5及TdT阳性。CD20和CD34表达程度不一,CD10可阴性。髓系标记阳性并不能除外B-ALL。CD79a和PAX5是最常用来表明B细胞分化的标记。但有的T-ALL患者可以表达CD79a,伴t(8;21)的AML的PAX5也可以阳性。抗MPO抗体免疫组化染色阴性,可以除外AML和B/My双表型AL。
B系原始淋巴细胞分化程度与临床和遗传学异常有关。Pro-B-ALL表达CD19、cCD79a、cCD22和TdT,而中间阶段即普通型ALL表达CD10,Pre-B-ALL则阳性。B-ALL通常不表达SIg。但SIg阳性,只要其他表型、形态和遗传特征符合,并不能除外B-ALL。
免疫表型分析也是区分正常B祖细胞与B-ALL治疗后微小残留病变(MRD)的重要手段。在流式细胞仪免疫表型分析图上,前者CD20等B细胞成熟标记表达呈由弱到强的连续分布,不同抗原之间表达是协调的。但在B-ALL细胞中,这些标记(如CD10、CD45、CD38、CD58和TdT等)的表达强度比较一致,或过强或过弱,图形上聚集成团,且不同抗原间表达不协调,呈絮乱之象。
(3)遗传学:几乎所有B-ALL患者IgH基因呈现DJ单克隆重排,70%的患者TCR也呈单克隆重排。遗传学异常有多种,常见的是6q、9p和12p缺失,但是对预后没有影响。t(17;19)和21号染色体上RUNX1基因扩增者占ALL的5%,此类患者预后不良。
2. ALL伴重现性遗传学异常
(1)ALL伴t(9;22)(q34.1;q11.2);BCR-ABL1
在儿童中发病率较低,占儿童ALL的2%~4%,随着年龄的增加,发病率也增加,占成人ALL的25%。本型在各年龄段的预后都是最差的。形态与其他类型ALL相同。典型的此类患者表达CD10、CD19和TdT,常同时表达髓系抗原CD13和CD33。一般不表达CD117。但至少在成人中CD25的表达与此类ALL高度密切相关。t(9;22)的ALL很少为T细胞表型。具有t(9;22)易位的患者的生存期明显短于t(9;22)易位阴性的患者。在儿童和成人中如果为t(9;22)易位阳性,应采取更积极的治疗措施,如HSCT。
(2)ALL伴t(v;11q23.3);KMT2A重排
主要见于1岁以内的婴儿,其他儿童较少见。在成人阶段发病又增加。临床上患者就诊时外周血WBC数常>100×109/L,易累及中枢神经系统。形态与其他类型ALL相似。CD19、CD15和NG2阳性,但CD10和CD24阴性。KMT2A通常与4q21的AF4、19p13的ENL和9p22的AF9发生易位。有些遗传学异常并非本型特异,如KMT2A-ENL也见于T-ALL,典型的KMT2A-AF9见于AML。FLT3常过度表达。具有11q23易位的患者预后极差,临床上归为高危组。
(3)ALL伴t(12;21)(p13.2;q22.1);ETV6-RUNX1
儿童常见,占儿童B-ALL的25%,但未在婴幼儿中见到,在较大儿童中发病也减少,成人罕见。临床表现、形态及细胞化学特征同其他类型ALL。原始细胞表达CD19和CD10,CD34也常阳性。CD9、CD20和CD66c阴性,这是相对特异性的特点。经常表达髓系抗原,尤其是CD13,但不意味着是混合白血病。融合蛋白ETV6-RUNX1以显性负调控的方式抑制转录因子RUNX1的功能。本型预后良好,儿童的治愈率>90%,尤其是当患者没有其他不良预后因素(如>10岁、WBC高等)时,复发较其他类型晚。
(4)ALL伴超二倍体核型
儿童常见,占B-ALL的25%。婴幼儿患者少见,发生率随年龄的增长而下降,成人很少发病。临床表现形态及细胞化学特征同其他类型ALL相似。原始细胞CD19和CD10阳性,多数CD34阳性,CD45常为阴性。白血病细胞染色体数>50条,通常<66条,一般无染色体易位和结构异常,无确定的染色体数目的限制。最常见的染色体异常是数目增加,发生率由高到低依次是21、X、14和4号,其次是1、2和3号。本型患者预后良好,治愈率>90%,尤其是4、10和17号染色体同时为三体者。
(5)ALL伴亚二倍体核型
白血病细胞染色体<46条,严格定义为<45甚至<44条染色体,可能更反映本病的本质。染色体为24-31条为近单倍体、32-39条为低亚二倍体、40-45条为近二倍体。此类白血病占所有ALL的5%,<45条者约占所有ALL的1%,儿童和成人均可见,但近单倍体(23~29条染色体)的患者主要见于儿童。临床表现、形态及细胞化学特征同其他类型ALL相似。CD19和CD10常阳性,无其他特殊表型。常规核型分析容易漏掉近单倍体或数目少的亚二倍体,亚二倍体B-ALL预后差。但相对而言,有44或45条染色体者预后最好,而近单倍体最差。
(6)ALL伴t(5;14)(q31.1;q32.3);IL3-IGH
在ALL中占<1%,儿童和成人均可有,临床上无特殊表现。形态上特别之处是嗜酸性粒细胞反应性增多,可能是IL-3产生过多所致。在BM原始细胞较低时,诊断可依据免疫分型和遗传学异常而定。原始细胞表达CD19和CD10。如果患者的原始细胞较少,但具有此类表型并发现嗜酸性粒细胞增多者强烈提示本病。
(7)ALL伴t(1;19)(q23;p13.3);TCF3-PBX1
占儿童ALL的6%,也可见于成人,但发病率低于儿童。典型表型为Pre-B-ALL,表达CD19、CD10和cμ,但不是所有的病例均为Pre-B-ALL表型。如果为cμ阳性,原始细胞强表达CD9,CD34一般为阴性,或者只有少数细胞表达低水平的CD34则提示为此类白血病。文献报道25%的病例具有t(1;19)(q23;p13.3)产生的TCF3-PBX1融合基因,本病的发生与TCF3-PBX1融合基因抑制正常转录因子E2A/TCF3和PBX1的功能相关。其临床表现、形态及细胞化学特征同其他类型ALL相似,本型需要除外t(17;19)和超二倍体时伴随的t(1;19)。前者预后差,后者易位的染色体虽然与本型完全相同,但涉及的基因不是E2A/TCF3和PBX1。
(8)暂定分型:BCR-ABL1样ALL(BCR-ABL1-like ALL)
对于此种类型AL的定义仍存在困难,2009年分别由两个研究组发现Ph阴性ALL的一种新型高危亚型,提出Ph样ALL的概念,其基因表达谱与BCR-ABL1阳性ALL类似,伴有IKZFl或其他淋巴转录调节因子缺失,临床预后也相似,为一组高危疾病,故被称为Ph样ALL(Ph 1ike ALL或BCR/ABL1-like ALL)。现已逐渐认识到Ph样ALL虽然基因组水平的异常具有显著的异质性,但共同特征主要是细胞因子受体和激酶信号通路活化相关的分子异常,同时常伴有淋系发育相关转录因子的异常。此种类型AL常提示预后不良,部分患者对TKIs治疗有效。BCR-ABL1样ALL共同特征是涉及其他酪氨酸激酶的易位、激酶受体样因子 2(CRLF2)易位,还包括红细胞生成素受体(EPOR)截短重排和激活等。CRLF2易位患者常与JAK基因突变有关。涉及酪氨酸激酶突变的易位可以累及ABL1基因(伙伴基因并非BCR基因)、ABL2、PDGFRB、NTRK3、TYK2、CSF1R和JAK2等,目前已报道30余种伴侣基因,其中EBF1-PDGFRB+ALL患者TKIs治疗效果较好。BCR-ABL1样ALL中IKZF1和CDKN2A/B缺失发生率较高,但是此种缺失在其他类型的ALL也可见到。
(9)暂定分型:伴21号染色体内部扩增的B-ALL(with intrachromosomal amplification of chromosome 21,iAMP21)(ALL伴iAMP21)
此型ALL占儿童ALL的2%,尤其是年龄较大的儿童,成年人少见。临床常见WBC数低。此型ALL主要是21号染色体内部扩增,通过FISH探针检测 RUNX1基因可以发现5个或者5个以上的基因拷贝,或中期分裂细胞的一条染色体上≥3个拷贝。研究发现,拷贝数的多数变化是以21号染色体为靶点,增加该染色体的复杂性。21号染色体的共有扩增区域为5.1Mb区,包括RUNX1、miR-802及定位于唐氏综合征临界区域的一些基因。通过基因组研究,影响关键通道基因的反复性异常得到了确认:IKZF1占22%、CDKN2A/B占17%、PAX5占8%、ETV6占19%和RB1占37%。克隆构型研究证实,这些异常以及P2RY8-CRLF2,都是在21号染色体重排后发生的。不论是否存在这些变化,患者的标准治疗结果均不佳。研究也表明21号染色体的不稳定性是iAMP21患者所共有的唯一的异常现象,因此,引发疾病的基因事件很可能隐藏在该异常染色体复杂的结构重排中。ALL伴iAMP21常提示预后差,但对于部分患者,强化疗可能有效。
3.T淋巴母细胞白血病
暂定分型:早期前体T淋巴母细胞白血病
暂时新增早期前体T淋巴母细胞白血病(ETP-ALL),此类型AL大多预后不良,但有部分研究得到可喜的治疗效果。其有特征性改变,如表达CD7和胞质CD3,CD4、CD2可以阳性。CD1a和CD8阴性,有1个或多个髓系或者干细胞抗原,如CD34、CD117、HLA-DR、CD13、CD33、CD11b或者CD65,CD5一般阴性,或阳性率<75%。常有髓系相关基因突变,如 FLT3、NRAS/KRAS、DNMT3A、IDH1和IDH2等;T-ALL基因常见突变,如 NOTCH1或CDKN1/2发生率较低。
4.暂定分型:自然杀伤(NK)细胞淋巴细胞白血病
常见于男性青少年,全血细胞减少较常见,常表达 CD56、CD16、CD2和CD7,部分CD8阳性,sCD3和CD4常阴性,无 TCRγδ及TCRαβ的表达。此类型AL表现凶险,进展快。
(六)鉴别诊断
1传染性单核细胞增多症:EB病毒感染所致的疾病,临床表现有发热、咽峡炎、浅表淋巴结及肝脾肿大,部分有皮疹,外周血淋巴细胞比例增高,异形淋巴细胞细胞升高超过10%。其中III型细胞胞体大,细胞核形态幼稚,易与原始淋巴细胞混淆。但此类患者骨髓及外周血没有原始淋巴细胞,血液嗜异凝集试验阳性,血清EB病毒抗体阳性,可与急性淋巴细胞白血病鉴别。
2 急性髄系白血病M0、M1及急性混合细胞白血病:临床表现及体征与急性淋巴细胞白血病相似,细胞形态亦难以区分,主要依据细胞表面抗原进行区分。
3 慢性粒细胞白血病急淋变:伴有Ph染色体或者/bcr-abl融合基因急性淋巴细胞白血病与部分以淋巴细胞急性变起病的慢性粒细胞白血病患者难以区分。一般而言,前者的融合产物多为P190,后者以P210更为常见。二者治疗反应亦不同。伴有Ph染色体或者/bcr-abl融合基因急性淋巴细胞白血病通过化疗获得完全缓解后往往能够获得细胞及分子遗传学的完全缓解,慢性粒细胞白血病急变的患者化疗缓解后通常回复至慢性期,获得细胞及分子遗传学的完全缓解罕见。
4 再生障碍性贫血及免疫性血小板减少症:二者血象与白细胞不增多的白血病可能混淆,但肝脾淋巴结不大,应注意骨髓形态学的特点(有无异常增多的白血病细胞)、染色体检查常无异常。
5 慢性淋巴细胞白血病及幼淋细胞白血病:二者均表现为淋巴细胞增高,可有肝脾、淋巴结肿大,但多数临床进展缓和,骨髓及外周血中以成熟淋巴细胞为主,后者幼稚淋巴细胞可超过55%。可通过细胞免疫表型分析与急性淋巴细胞白血病鉴别。
四、治疗
患者一经确诊后应尽快开始治疗,治疗应根据疾病分型采用合适的治疗方案、策略。
以下患者给予预治疗,以防止肿瘤溶解综合征的发生: ALL(Ph阴性或Ph阳性)患者,若WBC≥50×109/L,或者肝、脾、淋巴结肿大明显,或有发生肿瘤溶解特征的患者。预治疗方案:糖皮质激素(如泼尼松、地塞米松等)口服或静脉用,连续3~5 天。可以和环磷酰胺(CTX)联合应用(每天200 mg/m2,静脉滴注,连续3~5 天)。
(一)Ph阴性-ALL(Ph--ALL)的治疗
1.诱导治疗:
(1)治疗原则
年龄<40岁的患者:①临床试验;或②多药联合化疗(优先选择儿童特点方案)。
年龄≥40岁的患者:①<60岁的患者,可以入组临床试验,或采用多药联合化疗;②≥60岁者,可以入组临床试验,或采用多药化疗(不强调门冬酰胺酶的应用),或糖皮质激素诱导。
临床试验:如常规的、前瞻性系统治疗方案;CD20阳性的ALL患者可以采用化疗联合抗CD20单克隆抗体的治疗方案;其他有科学依据的探索性研究方案等。
(2)具体治疗方案组合
一般以4周方案为基础。至少应予长春新碱(VCR)或长春地辛、蒽环/蒽醌类药物[如柔红霉素(DNR)、去甲氧柔红霉素(IDA)、阿霉素、米托蒽醌等]、糖皮质激素(如泼尼松、地塞米松等)为基础的方案(VDP)诱导治疗。推荐采用VDP联合CTX和门冬酰胺酶(L-Asp)组成的VDCLP方案,鼓励开展临床研究。
也可以采用Hyper-CVAD方案。
诱导治疗中:
①蒽环/蒽醌类药物:可以连续应用(连续2-3 d,第1、3周,或仅第1周用药);也可以每周用药1次。用药参考剂量:DNR 30~45 mg/(m2·d)×2~3 d,IDA 6~10 mg/(m2·d)×2~3 d,米托蒽醌(Mitox)6~10 mg/(m2·d)×2~3 d。
②单次应用CTX剂量较大时(超过1 g)可以予美司钠解救。
③诱导治疗第14天复查骨髓,根据骨髓情况调整第3周的治疗。诱导治疗第28(±7)天判断疗效,未能达CR的患者进入挽救治疗。
④尽早开始腰穿、鞘注,预防中枢神经系统白血病(CNSL)(可选择在血细胞计数安全水平时进行)。
2.CR后的治疗:为减少复发、提高生存率,诱导治疗结束后应尽快开始缓解后的巩固强化治疗。应根据患者的危险度分组情况判断是否需要行异基因造血干细胞移植(allo-HSCT),需行allo-HSCT者积极寻找供者。
(1)治疗原则
年龄<40岁的患者:①继续多药联合化疗(尤其是微小残留病MRD阴性者);或②allo-HSCT (尤其是MRD阳性,高白细胞计数患者,伴预后不良细胞遗传学异常的B-ALL、T-ALL)。
年龄≥40岁的患者:①<60岁的患者,继续多药联合化疗(尤其是MRD阴性者);或考虑allo-HSCT (尤其是MRD阳性,高白细胞计数患者,伴预后不良细胞遗传学异常的B-ALL、T-ALL)。②≥60岁的患者或不适合强烈治疗者(高龄、体能状态较差、严重脏器并发症等)可考虑继续化疗。
(2)具体注意事项
缓解后强烈的巩固治疗可清除残存的白血病细胞、提高疗效,但是巩固治疗方案在不同的研究组、不同的人群并不相同。一般应给予多疗程的治疗,药物组合包括诱导治疗使用的药物(如长春碱类药物、蒽环类药物、糖皮质激素等)、大剂量甲氨蝶呤(HD-MTX)、阿糖胞苷(Ara-C)、6-巯嘌呤(6-MP)、门冬酰胺酶等。因此,缓解后治疗可以有1~2个疗程再诱导方案,2~4个疗程HD-MTX、Ara-C、L-Asp的方案。
在整个治疗过程中应强调参考儿童ALL方案的设计,强调非骨髓抑制性药物(包括糖皮质激素、长春碱类、L-Asp)的应用。
a.一般应含有HD-MTX方案。MTX 1~3.0 g/m2(T-ALL 可以用到5 g/m2)。应用HD-MTX时应争取进行血清MTX浓度监测,注意甲酰四氢叶酸钙的解救,至血清MTX浓度<0.1 μmol/L(或低于0.25 μmol/L)时结合临床情况可停止解救。
b.应含有Ara-C为基础的方案。Ara-C可以为标准剂量、分段应用(如CTX、Ara-C、6-巯嘌呤为基础的方案),或中大剂量Ara-C为基础的方案。
c.可以继续应用含L-Asp的方案(大肠杆菌或欧文氏菌来源,或培门冬酶)。
d.缓解后6个月左右参考诱导治疗方案予再诱导强化一次。
e.干细胞移植的问题:考虑allo-HSCT的患者应在一定的巩固强化治疗后尽快移植。无合适供者的高危组患者(尤其是MRD持续阴性者)、标危组患者(MRD阴性者)可以考虑在充分的巩固强化治疗后进行AHSCT。AHSCT后的患者应继续予一定的维持治疗。
无移植条件的患者、持续属于低危组的患者按计划巩固强化治疗。
3.维持治疗:ALL患者强调维持治疗,维持治疗的基本方案: 6-MP 60~75 mg/m2每日1次,MTX 15~20 mg/m2每周1次。注意:①6-MP晚上用药效果较好。可以用硫鸟嘌呤(6-TG)替代6-MP。维持治疗期间应注意监测血常规和肝功能,调整用药剂量。②ALL的维持治疗既可以在完成巩固强化治疗之后单独连续使用,也可与强化巩固方案交替序贯进行。③自取得CR后总的治疗周期至少2年。
维持治疗期间应尽量保证每3~6个月复查1次。
推荐方案:
1.中国成人急性淋巴细胞白血病协作组(CALLG)——CALLG-2008治疗方案。
2.CALGB8811方案(Larson RA. Blood,1995,85:2025-2037)。
3.BFM强化方案(Stock W. Blood,2008,112:1646-1654)。
4.Hyper-CVAD方案(MDACC)(Kantarjian H. Cancer,2004,101:2788-801)。
5.MRC UKALLXII/ECOG E2993(Rowe JM. Blood,2005,106: 3760-3767)。
6.DFCI Pediatric ALL Consortium regimen(DeAngelo DJ. Leukemia,2015,29: 526–534)。
7.ALL IC-BFM 2002(Star J. J Clin Oncol,2013,32:174-184)。
(二)Ph阳性-ALL(Ph+-ALL)的治疗
1.非老年(年龄<60岁的患者)Ph+-ALL的治疗
(1)诱导缓解治疗:①临床试验。②多药化疗+酪氨酸激酶抑制剂(TKI)治疗。
诱导治疗和一般Ph阴性-ALL一样,建议予VCR或长春地辛、蒽环/蒽醌类药物、糖皮质激素为基础的方案(VDP)诱导治疗;鼓励进行临床研究。
一旦融合基因(PCR方法)或染色体核型/荧光原位杂交(FISH)证实为Ph/BCR-ABL1阳性ALL则进入Ph+-ALL治疗序列,可以不再应用L-Asp。自确诊之日起即可以加用(或酌情于第8或15天开始)TKI,推荐用药剂量:伊马替尼400~600 mg/d、达沙替尼100-140 mg/d;优先推荐TKI持续应用。若粒细胞缺乏(尤其是中性粒细胞绝对值<0.2×109/L)持续时间较长(超过1周)、出现感染发热等并发症时,可以临时停用TKI,以减少患者的风险。
诱导治疗第14天复查骨髓,根据骨髓情况调整第3周的治疗。诱导治疗第28(±7)天判断疗效,同时复查骨髓和细胞遗传学(诊断时有异常者)、BCR-ABL融合基因,判断疗效。有造血干细胞移植条件者,行HLA配型,寻找供者。
尽早开始腰椎穿刺、鞘内注射,预防CNSL(可选择在血细胞计数安全水平时进行)。
(2)CR后的治疗
Ph+-ALL的缓解后治疗原则上参考一般Ph--ALL,但可以不再使用L-Asp。TKI优先推荐持续应用,至维持治疗结束(无条件应用TKI的患者按一般ALL的治疗方案进行)。
①有合适供者的患者可以选择allo-HSCT,移植后可以用TKI维持。
②无合适供者的患者,按计划继续多药化疗+TKI。
③无合适供者、BCR-ABL融合基因转阴性者(尤其是3-6个月内转阴性者),可以考虑自体造血干细胞移植(AHSCT),移植后予TKI维持。
④应定期监测BCR-ABL融合基因表达,CNSL的预防治疗参考一般ALL患者。
(3)维持治疗
①可以应用TKI治疗者,用TKI为基础的维持治疗(可以联合VCR、糖皮质激素,或6-MP和MTX;或联合干扰素),至CR后至少2年。
②不能坚持TKI治疗者,采用干扰素维持治疗,300万U/次,隔日1次 [可以联合VCR、糖皮质激素和(或)6-MP、MTX],缓解后至少治疗2年。或参考Ph--ALL进行维持治疗。
维持治疗期间应尽量保证每3~6个月复查1次:骨髓象、融合基因(BCR-ABL)定量和(或)流式细胞术残留病。
2.老年Ph+-ALL(年龄≥60岁)的治疗:老年Ph+-ALL的治疗原则上参考一般老年Ph--ALL,同时联合TKI。TKI优先推荐持续应用,至维持治疗结束。
(1)诱导治疗:①临床试验;②TKI+糖皮质激素;③TKI+多药化疗。
(2)CR后的治疗:继续TKI+糖皮质激素,或TKI+化疗巩固。之后参考非老年患者的维持治疗方案进行维持治疗。
推荐方案:
1.GMALL 06//99和07/03方案(Wassmann B,Blood,2006,108:1469-1477)。
2.Hyper-CVAD方案联合伊马替尼或达沙替尼(Thomas DA. Blood,2004,103: 4396-4407; Ravandi F. Blood,2010,116:2070–2077)(MDACC)。
3.Northern Italy Leukemia Group Protocol 09/00(Bassan R. J Clin Oncol,2010,28:3644-3652)。
4.JALSG ALL202(Yanada M. Br J Haematol,2008,143: 503-510)。
5.GIMEMA LAL0201-B(Vignetti M. Blood,2007,109:367)。
五、微小残留病(MRD)的监测
(一)MRD监测的时机
ALL整个治疗期间应强调规范的MRD监测,并根据MRD监测结果进行危险度和治疗调整。
1.早期——诱导治疗期间(第14天)和(或)结束时(第28天左右)。
2.缓解后定期监测,应保证治疗第16、22周左右的MRD监测。
早期的MRD检测主要用于预后的预测。缓解后MRD水平高的患者具有较高的复发危险,应进行较强的缓解后治疗,以改善长期疗效。
(二)MRD的监测方法
1.经典的MRD检测技术:①IG-TCR的定量PCR检测(DNA水平);②4-6色的流式细胞术MRD检测;③融合基因转录本的实时定量PCR(如BCR-ABL)。
2.新的高通量MRD检测技术:①基于EuroFlow的≥8色的二代流式细胞术MRD检测;②IG-TCR的高通量测序。
(三)Ph+-ALL疾病反复时应注意进行ABL激酶突变的分析。
六、中枢神经系统白血病(CNSL)的诊断、预防和治疗
CNSL是急性白血病(尤其是ALL)复发的主要根源之一,严重影响白血病的疗效。诊断时有中枢神经系统症状者应先进行物理检查(CT或核磁检查),排除出血或占位后再考虑腰椎穿刺,无神经系统症状者按计划进行CNSL的预防。
(一)CNS状态分类
CNS-1:白细胞分类无原始淋巴细胞(不考虑脑脊液白细胞计数)。
CNS-2:脑脊液白细胞计数<5个/ ml,可见原始淋巴细胞。
CNS-3:脑脊液白细胞计数≥5个/ml,可见原始淋巴细胞。
(二)CNSL诊断标准
目前CNSL尚无统一诊断标准。1985年在罗马讨论关于ALL预后差的危险因素时提出CNSL下列诊断标准:脑脊液白细胞计数≥0.005×109/L(5个/ml),离心标本证明细胞为原始细胞者,即可诊断CNSL。
流式细胞术检测脑脊液在CNSL中的诊断意义尚无一致意见,但出现阳性应按CNSL对待。
(三)CNSL的预防
任何类型的成人ALL均应强调CNSL的早期预防。预防措施可以包括:①鞘内化疗;②放射治疗;③大剂量全身化疗;④多种措施联合。
1.鞘内化疗:诱导治疗过程中没有中枢神经系统症状者可以在血细胞计数安全水平后行腰椎穿刺、鞘内注射(如PLT≥50×109/L)。鞘内注射主要用药包括地塞米松、MTX、Ara-C。常用剂量为MTX 10~15mg/次、Ara-C(30~50mg/次)、地塞米松三联(或两联)用药。
巩固强化治疗中也应进行积极的CNSL预防,主要是腰椎穿刺、鞘内注射(鞘内注射次数一般应达6次以上,高危组患者可达12次以上),鞘内注射频率一般不超过2次/周。
2.预防性头颅放疗:18岁以上的高危组患者或35岁以上的患者可进行预防性头颅放疗,放疗一般在缓解后的巩固化疗期或维持治疗时进行。预防性照射部位为单纯头颅,总剂量1 800~2 000 cGy,分次完成。
(四)CNSL的治疗
确诊CNSL的患者,尤其是症状和体征较明显者,建议先行腰椎穿刺、鞘内注射:MTX(10-15 mg/次)+Ara-C (30-50 mg/次)+地塞米松三联(或两联),每周2次,直至脑脊液正常;以后每周1次×4-6周。
也可以在鞘注化疗药物至脑脊液白细胞数正常、症状体征好转后再行放疗(头颅+脊髓放疗)。建议头颅放疗剂量2000-2 400 cGy、脊髓放疗剂量1 800-2 000 cGy,分次完成。进行过预防性头颅放疗的患者原则上不进行二次放疗。
七、ALL治疗反应的定义
(一)骨髓和外周血疗效标准
1. CR:①外周血无原始细胞,无髓外白血病;②骨髓三系造血恢复,原始细胞<5%;③中性粒细胞绝对计数(ANC)>1.0×109/L;④PLT>100×109/L;⑤4周内无复发。
2.CR伴血细胞不完全恢复(CRi):PLT<100×109/L和(或)ANC<1.0×109/L。其他应满足CR的标准。总反应率(ORR)=CR+CRi。
3.难治性疾病:诱导治疗结束未能取得CR。
4.疾病进展(PD):外周血或骨髓原始细胞绝对数增加25%,或出现髓外疾病。
5.疾病复发:已取得CR的患者外周血或骨髓又出现原始细胞(比例>5%),或出现髓外疾病。
(二)CNS疾病的治疗反应
1.CNS缓解:CNS-2或CNS-3患者取得CNS-1状态。
2.CNS复发:发生CNS-3状态或出现CNS白血病的临床症状(如面神经麻痹、脑/眼受累,或下丘脑综合征表现)。
(三)纵隔疾病的治疗反应
纵隔疾病的疗效判断依赖胸部CT和PET-CT。CR:CT检查纵隔肿大完全消失;或PET阴性。PR:肿大的纵隔最大垂直直径的乘积(SPD)缩小50%以上。PD:SPD增加25%以上。NR:不满足PR或PD。复发:取得CR的患者又出现纵隔肿大。
责任编辑: 夢奕新



 苏公网安备 32011402010112号
苏公网安备 32011402010112号